Host-Pathogen Investigation
Our lab has applied genetic tools to uncover how viruses exploit mammalian proteins to further their goals of reproduction and how the cell combats these viruses. We identified a large number of host factors required for a wide variety of viruses to replicate and factors that restrain this replication. We hope that by understanding these pathways, we can inform antiviral drug discovery and host-pathogen biology. We have also initiated a chemical screening effort to explore how existing FDA approved drugs can be used in combination to control acute viral infections.
- Recent Studies
- The Human Virome Project
- VirScan: comprehensive serological profiling of human populations using a synthetic human virome
- Measles virus diminishes preexisting immunological memory that offers protection from other pathogens
- Systematic autoantigen analysis identifies a distinct subtype of scleroderma with coincident cancer
- Viral epitope profiling of COVID-19 patients reveals cross-reactivity with common coronaviruses and correlates of disease severity
- Older Work
Recent Studies
The Human Virome Project

Emerging pathogens pose a world-wide threat as evidenced by multiple pandemics that have engulfed humanity in the last century alone. Human pathogens are viral, bacterial, protozoan or fungal in nature. Their zoonotic transmission to humans is increasing due to human encroachment upon animal habitats and the effects of climate change allowing infectious diseases to expand their range. Of these threats, potentially the most dangerous are viruses. Modern interconnectivity allows a single zoonotic transmission to circle the globe in a matter of weeks before it can even be firmly confirmed as a new virus. Many viruses exist in animal reservoirs and those that successfully transfer to humans are related to families of existing viral pathogens in humans such as influenza, coronaviruses, enteroviruses, picornaviruses, paramyxoviruses, flaviviruses, ebolavirus, retroviruses, and others, but in principle members of any viral family can evolve to be threats to humanity. For most of history, viral research has been siloed in individual labs for individual viruses with the exceptions of modern epidemics like HIV, Zika and SARS-CoV-2. For most viral proteins, we know very little about their individual functions – information that can be extremely valuable for combating viral diseases.
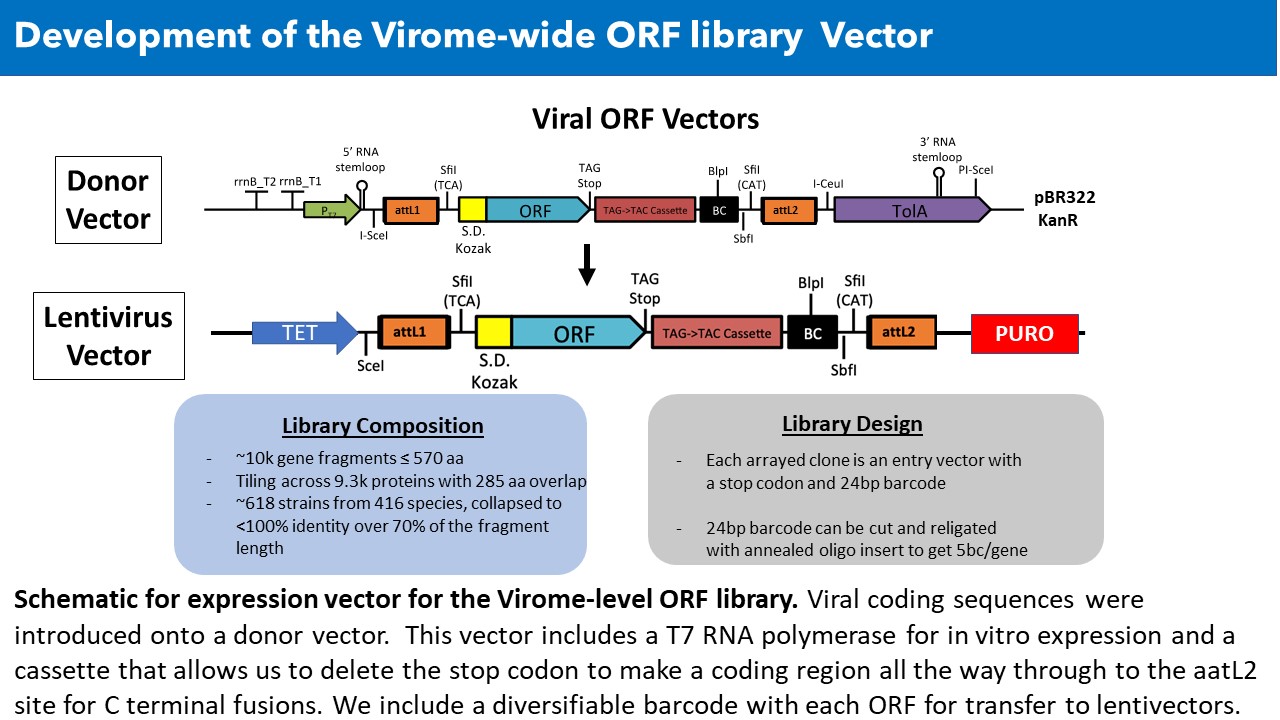
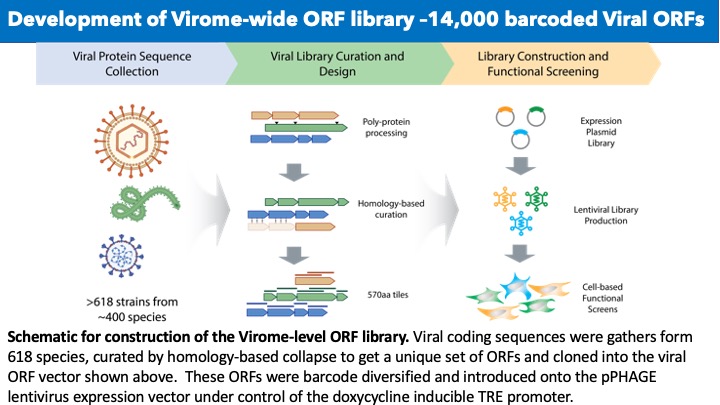
Viruses reprogram host cells to execute their programs of replication, immune evasion and transmission using a relatively small number of highly evolved proteins whose every surface is sculpted for specific activities. However, the functions of most viral proteins have not been studied with uniform assays. We feel viral research needs a formal systematic foundation of information to understand the strategies viruses implement and the proteins that virus’s impact to execute their strategies. Thus, we have set out to establish a platform to establish a foundation of knowledge for nearly all proteins from all known human viruses. We have already assembled a library of over 8000 arrayed viral proteins (vORFs) in barcoded vectors that are capable of expression in any format for genetic, cell biological or biochemical studies. We plan to expand this to ~20,000 vORFs and to explore vORF functions by performing high through-put analyses. We seek to create an atlas describing the full suite of mechanisms viruses use to hijack hosts to enable their replication and immune evasion.
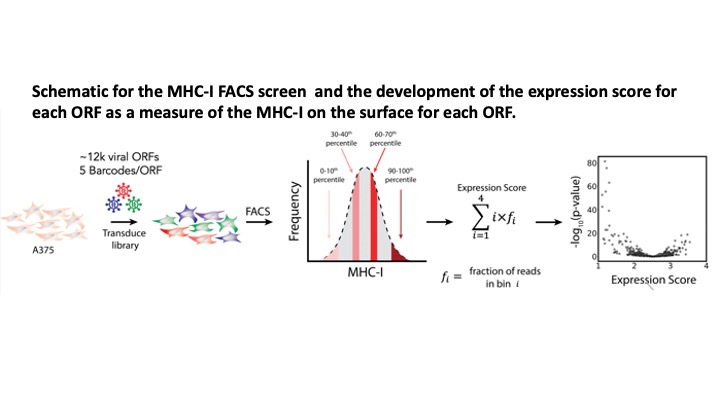
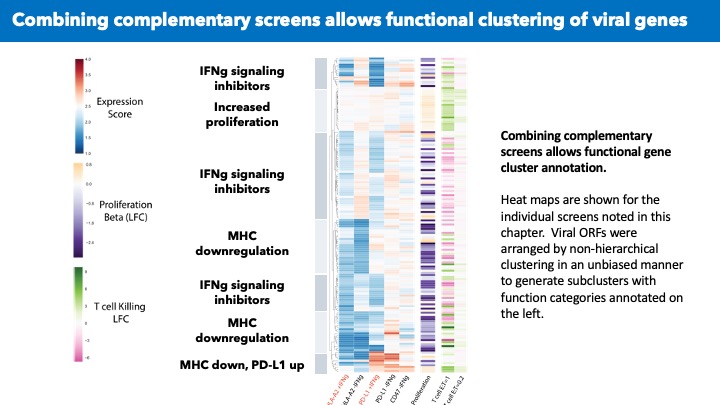
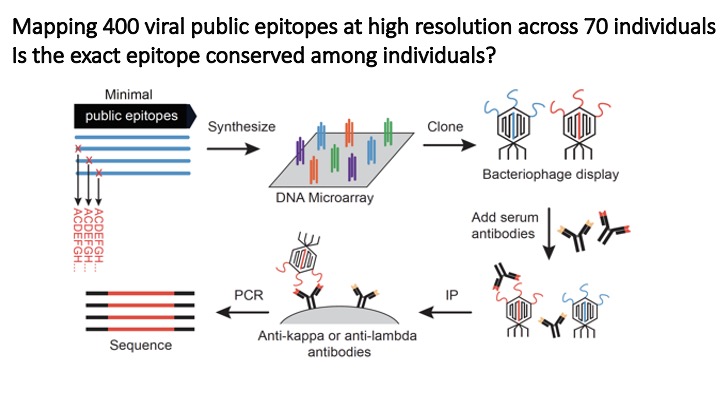
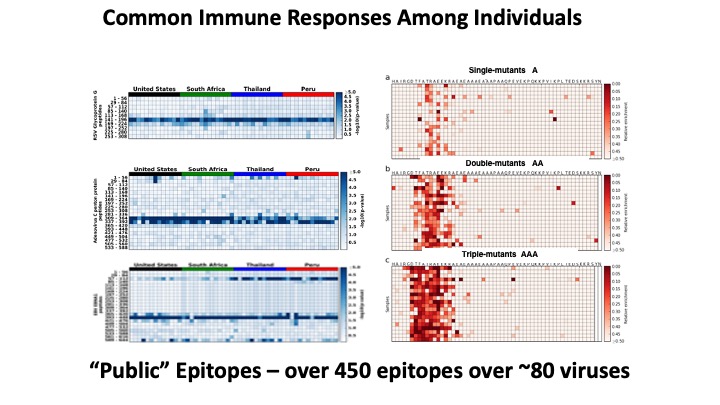
VirScan: comprehensive serological profiling of human populations using a synthetic human virome
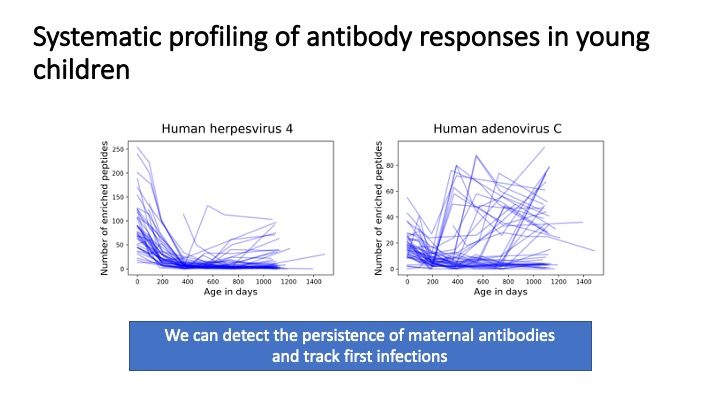
The human virome plays important roles in health and immunity. However, current methods for detecting viral infections and antiviral responses have limited throughput and coverage. To address this need we developed VirScan, a high-throughput method to comprehensively analyze antiviral antibodies using immunoprecipitation and massively parallel DNA sequencing of a bacteriophage library displaying proteome-wide peptides from all human viruses. We assayed over 108 antibody-peptide interactions in 569 humans across four continents, nearly doubling the number of previously established viral epitopes. We detected antibodies to an average of 10 viral species per person and 84 species in at least two individuals. Although rates of specific virus exposure were heterogeneous across populations, antibody responses targeted strikingly conserved “public epitopes” for each virus, suggesting that they may elicit highly similar antibodies. VirScan is a powerful approach for studying interactions between the virome and the immune system.

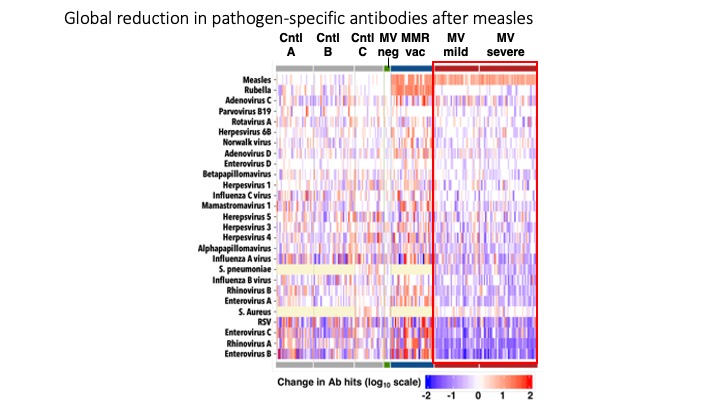
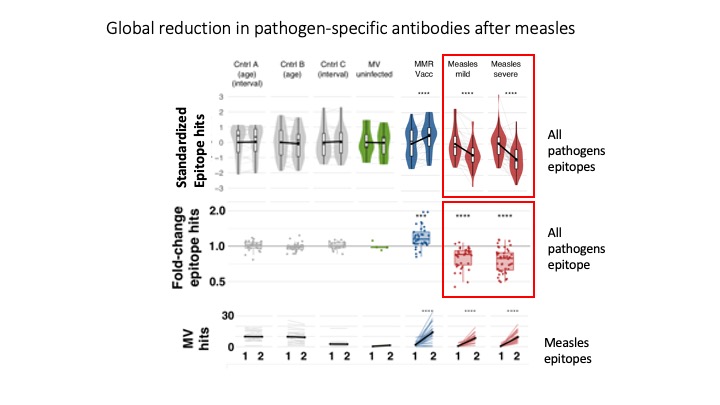
Measles virus diminishes preexisting immunological memory that offers protection from other pathogens
Measles causes over 100,000 deaths yearly. Epidemiological associations suggest far greater mortality stretching over years, but how this occurs was not understood. Measles virus infects immune cells producing acute immune suppression, but unexamined post-recovery effects could persist long-term. To address this outstanding issue, we employed VirScan to track antibodies to 1,000s of pathogen epitopes before and after measles virus infection. We find that measles deletes 25-50% of the antibody repertoire diversity and severely diminishes the remaining 50-75%, restructuring the immune system causing immune-amnesia. These results replicate in measles virus-infected macaques. No evidence of similar effects in MMR-immunized infants was detected. Thus, the significant deletion of humoral immune memory following measles suggests a prolonged vulnerability with significant potential for future morbidity and mortality and underscores the need for widespread vaccination.
Systematic autoantigen analysis identifies a distinct subtype of scleroderma with coincident cancer
Scleroderma is a chronic autoimmune rheumatic disease associated with widespread tissue fibrosis and vasculopathy. Approximately two-thirds of all patients with scleroderma present with three dominant autoantibody subsets. Here, we used a pair of complementary high-throughput methods for antibody epitope discovery to examine patients with scleroderma with or without known autoantibody specificities. We identified a specificity for the minor spliceosome complex containing RNA Binding Region (RNP1, RNA recognition motif) Containing 3 (RNPC3) that is found in patients with scleroderma without known specificities and is absent in unrelated autoimmune diseases. We found strong evidence for both intra- and intermolecular epitope spreading in patients with RNA polymerase III (POLR3) and the minor spliceosome specificities. Our results demonstrate the utility of these technologies in rapidly identifying antibodies that can serve as biomarkers of disease subsets in the evolving precision medicine era.
Viral epitope profiling of COVID-19 patients reveals cross-reactivity with common coronaviruses and correlates of disease severity
Understanding humoral responses to severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is critical for improving diagnostics, therapeutics, and vaccines.
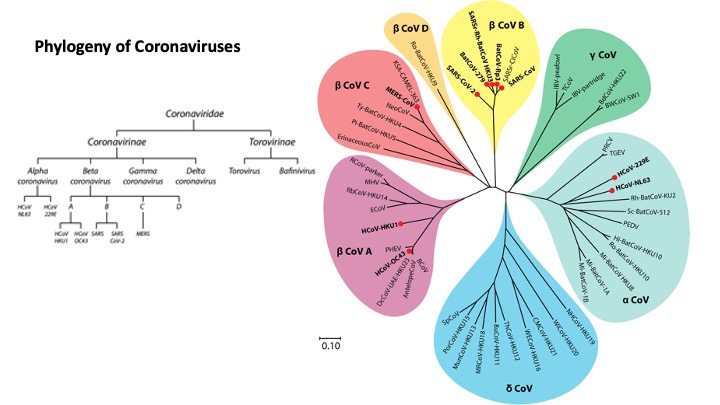
Deep serological profiling of 232 coronavirus disease 2019 (COVID-19) patients and 190 pre-COVID-19 era controls using VirScan revealed more than 800 epitopes in the SARS-CoV-2 proteome, including 10 epitopes likely recognized by neutralizing antibodies. Preexisting antibodies in controls recognized SARS-CoV-2 ORF1, whereas only COVID-19 patient antibodies primarily recognized spike protein and nucleoprotein. A machine learning model trained on VirScan data predicted SARS-CoV-2 exposure history with 99% sensitivity and 98% specificity; a rapid Luminex-based diagnostic was developed from the most discriminatory SARS-CoV-2 peptides. Individuals with more severe COVID-19 exhibited stronger and broader SARS-CoV-2 responses, weaker antibody responses to prior infections, and higher incidence of cytomegalovirus and herpes simplex virus 1, possibly influenced by demographic covariates. Among hospitalized patients, males produce stronger SARS-CoV-2 antibody responses than females.

Older Work
Most antiviral drugs target proteins on the virus. While these can have efficacy, the virus has a ready mechanism through which to overcome these drugs by mutating residues involved in drug binding. However, drugs that interfere with host pathways upon which the virus relies cannot be easily overcome by the virus by mutation because it would require the virus to evolve an entirely different functionality, and extremely low probability event. In order to identify potential anti-drug targets, we began an analysis of several important viral pathogens to identify the host factors upon which they rely with an eye toward finding anti-viral drug targets.
HIV: A Genetic Map of the HIV Life Cycle and Host Cell Dependencies
HIV-1 exploits multiple host proteins during infection. We performed the first large-scale genome-wide siRNA screen to identify host factors required by HIV-1 and identified 274 HIV-dependency factors (HDFs) (207).
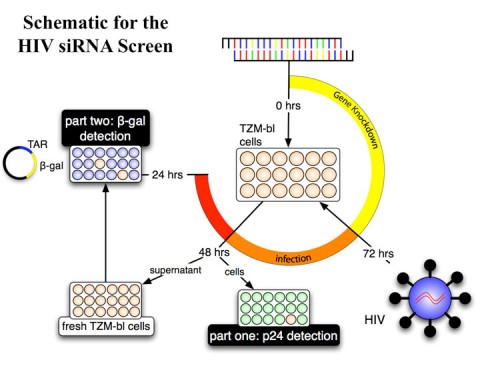
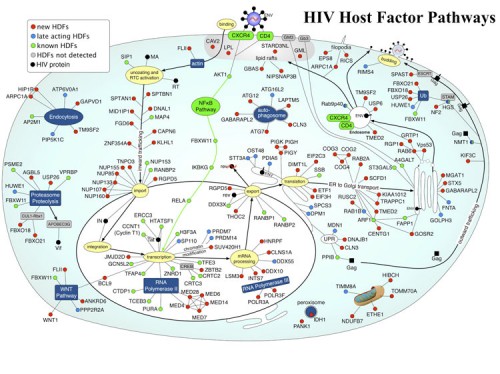
These proteins participate in a broad array of cellular functions and implicate new pathways in the viral lifecycle. Further analysis revealed previously unknown roles for retrograde Golgi transport proteins (Rab6 and Vps53) in viral entry, a karyopherin (TNPO3) in viral integration, and the Mediator complex (Med28) in viral transcription. Transcriptional analysis revealed that HDF genes were enriched for high expression in immune cells suggesting that viruses evolve in host cells that optimally perform the functions required for their lifecycle. These host factors represent potential therapeutic targets. We have mapped these functions onto a cell to show the pathways HIV must exploit to propagate. We are currently completing this work with funding from the Gates Foundation to use all existing RNA interference methods to identify all detectible host factors needed for the HIV lifecycle. This information will be integrated into protein interaction maps to realize an integrated view of the HIV lifecycle and possible points of intervention.
HCV: A Screen for Host Factors Needed for Viral Propagation
Hepatitis C virus (HCV) infection is a major cause of end stage liver disease and a leading indication for liver transplantation. Current therapy fails in many instances and is associated with significant side effects. HCV encodes only a few proteins and depends heavily on host factors for propagation. Each of these host dependencies is a potential therapeutic target. To find host factors required by HCV, we completed a genome-wide small interfering RNA (siRNA) screen using an infectious HCV cell culture system (232). We applied a two-part screening protocol to allow identification of host factors involved in the complete viral lifecycle.
The candidate genes found included known or previously identified factors, and also implicate many additional host cell proteins in HCV infection. To create a more comprehensive view of HCV and host cell interactions, we performed a bioinformatic meta-analysis that integrates our data with those of previous functional and proteomic studies. The identification of host factors participating in the complete HCV lifecycle will both advance our understanding of HCV pathogenesis and illuminate novel therapeutic targets.
Influenza A: Host Dependency Factors
Influenza viruses exploit host cell machinery to replicate, resulting in epidemics of respiratory illness. In turn, the host expresses anti-viral restriction factors to defend against infection. To find host-cell modifiers of influenza A H1N1 viral infection, we used a functional genomic screen and identified over 120 influenza A virus-dependency factors (IDFs) with roles in endosomal acidification, vesicular trafficking, mitochondrial metabolism, and RNA splicing (233).

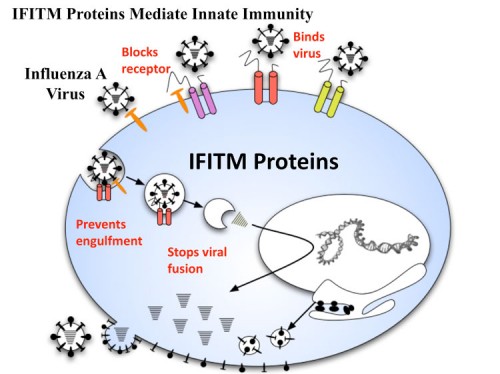
IFITM Proteins – Mediators of Antiviral Innate Immunity
The siRNA screen also led to the discovery that the interferon-inducible trans-membrane proteins, IFITM1, 2 and 3, restrict early replication of influenza A virus (233). The IFITM proteins confer basal resistance to influenza A virus, but are also inducible by interferons (IFN) type I and II, and are critical for IFN’s virustatic actions. Further characterization revealed that the IFITM proteins inhibit the early replication of flaviviruses, including dengue virus (DNV) and West Nile virus (WNV).
The IFITM protein family is highly conserved in evolution all the way down to fish. While it remains to be seen if it functions to control innate immunity in these organisms, the fact that the gene family is ancient and still works today to thwart influenza A and other viruses suggests that it acts in a manner not easily overcome by these viruses and is probably attaching a part of the pathway that does not involved recognizing the virus itself, which would allow a simple mechanism of circumvention by the virus, perhaps by rerouting its transport within the cells once it has been endocytosed. Our current clues on the function of these proteins are that they are expressed on the cell surface and internally on vesicles and that they work at entry as the first line defense against viral entry. Understanding the mechanism of their action should provide greater insight into how to combat Influenza and other viruses. In addition, variation in the levels or activity of IFITM proteins may lead to enhanced resistance or sensitivity to infection by the flu and the severity of the infection for individuals. Since removing IFITM proteins allows influenza A to replicate much more efficiently, we envision engineering vaccine production strains to inhibit IFITM proteins so as to speed up vaccine production. In addition, one of the major threats to human health is the large reservoir of influenza that exists in farm animals such as swine and poultry that have a large amount of human contact. The engineering of these animals to express higher levels of IFITM proteins could eliminate them as reservoirs of virus that then recombine with human-tropic viruses to create pandemic strains of flu.